Pompe Disease Life Expectancy With Treatment
For individuals diagnosed with Pompe disease, a rare and often debilitating genetic disorder, the question of life expectancy is paramount. While historically a diagnosis of Pompe disease, particularly in its infantile-onset form, carried a grim prognosis, advancements in treatment, most notably enzyme replacement therapy (ERT), have dramatically altered the landscape and offer hope for significantly extended lifespans. This article examines the evolving life expectancy for individuals with Pompe disease in the context of available treatments.
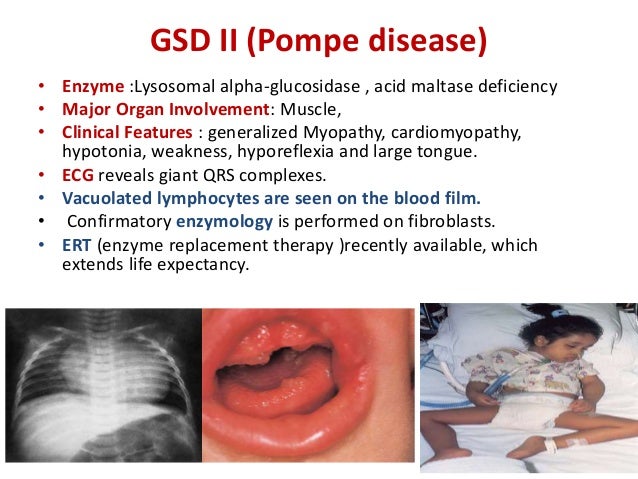
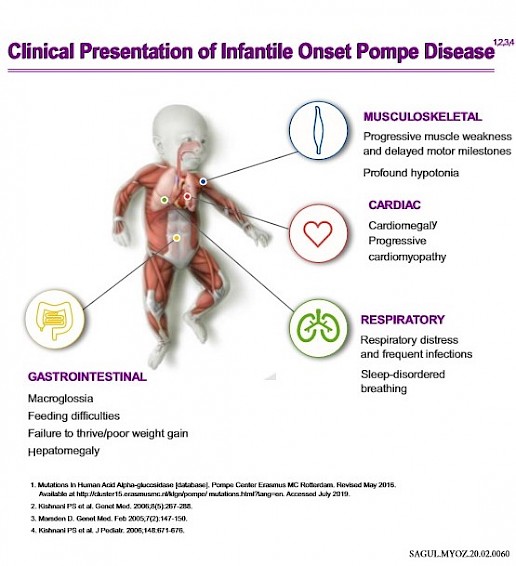
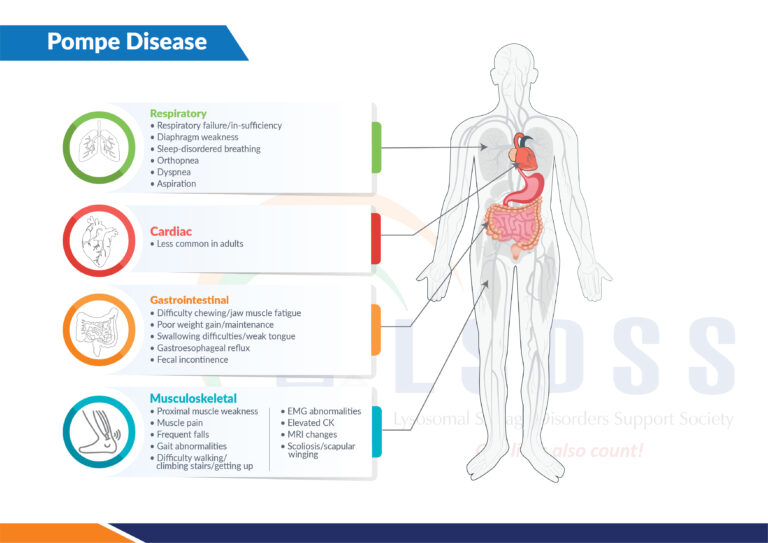
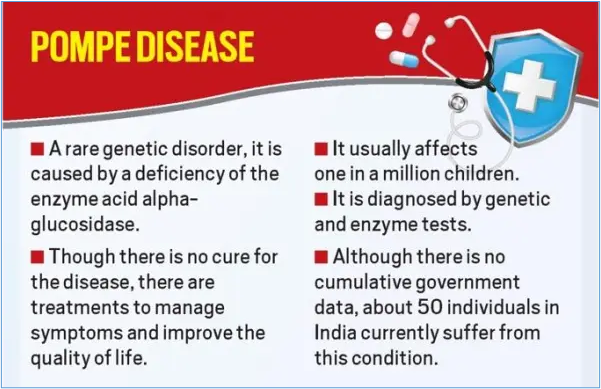
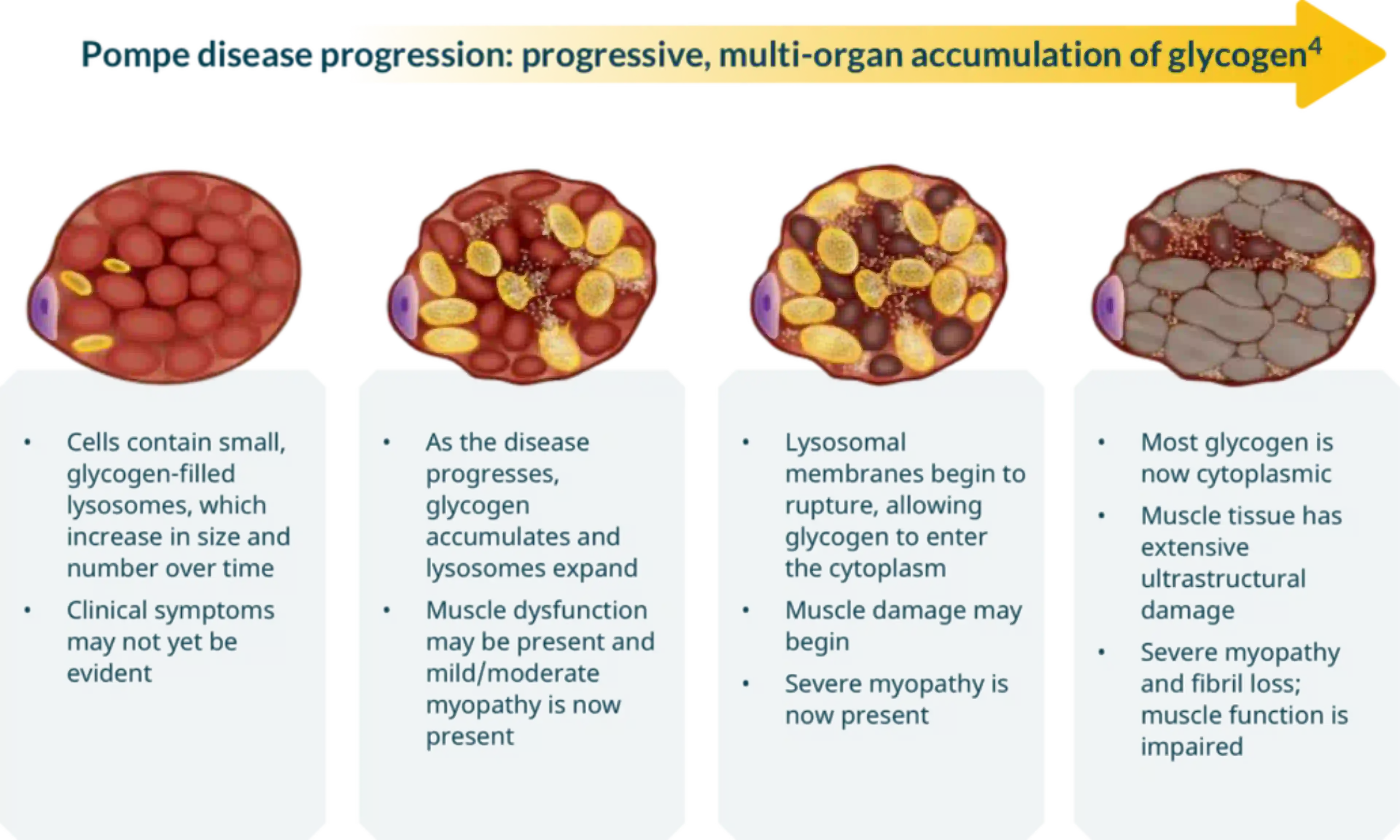
Pompe disease, caused by a deficiency in the enzyme acid alpha-glucosidase (GAA), leads to the buildup of glycogen in various tissues, primarily affecting the heart and skeletal muscles. This accumulation results in progressive muscle weakness, respiratory problems, and, in severe cases, premature death. Understanding the impact of treatment on life expectancy requires distinguishing between the different forms of Pompe disease and the timing of intervention.
Understanding Pompe Disease and Its Impact
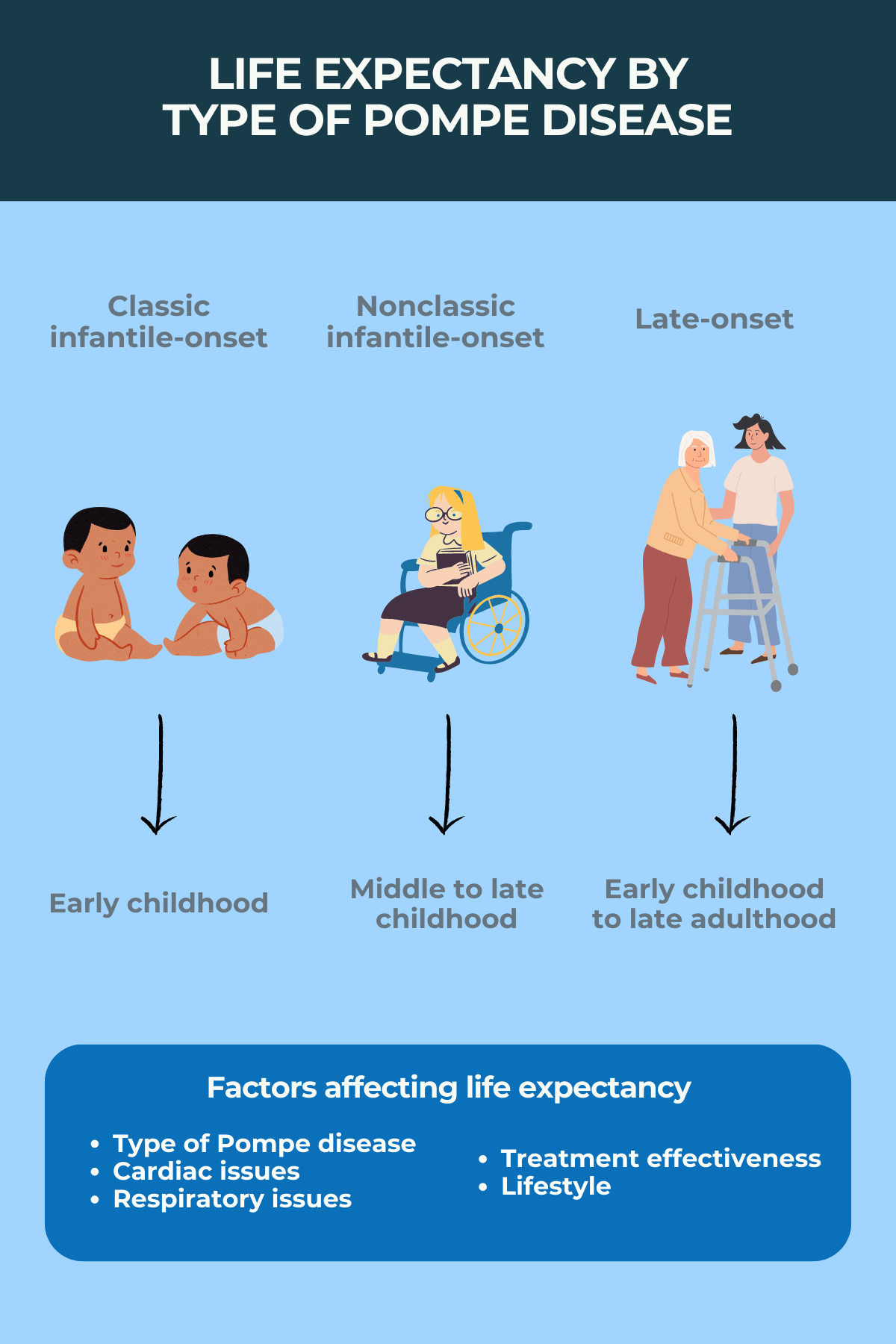
Pompe disease is classified into two main forms: infantile-onset and late-onset. Infantile-onset Pompe disease (IOPD) is the most severe form, typically presenting within the first few months of life.
Without treatment, infants with IOPD often succumb to cardiorespiratory failure within the first year. Late-onset Pompe disease (LOPD) manifests later in life, from childhood to adulthood, and progresses more slowly, but still leads to significant disability and reduced lifespan.
The Significance of Enzyme Replacement Therapy
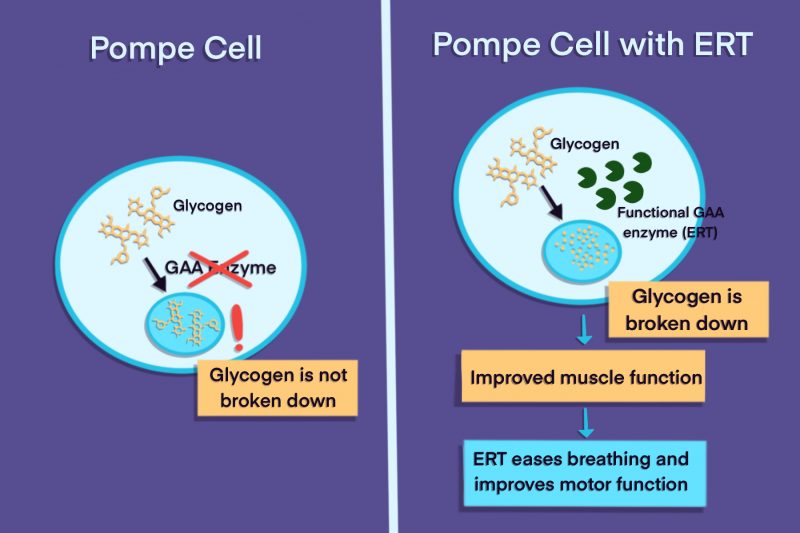
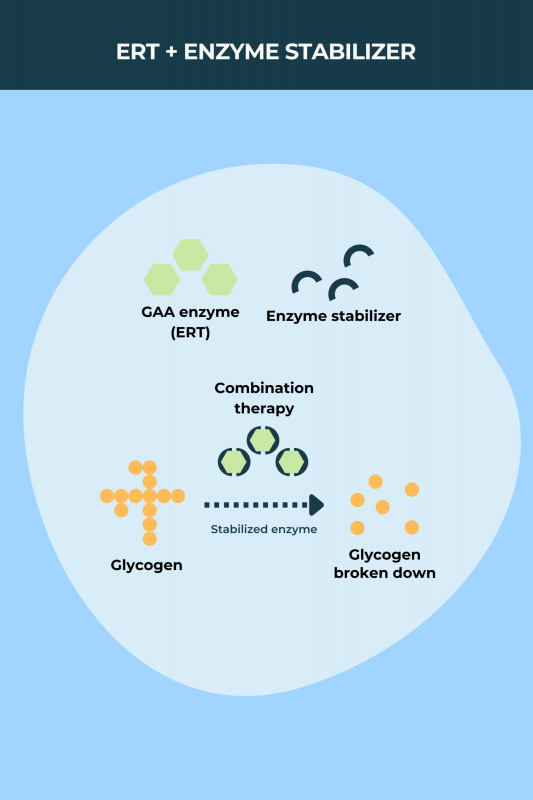
The introduction of enzyme replacement therapy (ERT) has revolutionized the treatment of Pompe disease. ERT involves intravenous infusions of a synthetic version of the GAA enzyme, aiming to reduce glycogen accumulation and improve muscle function.
Myozyme (alglucosidase alfa), the first approved ERT for Pompe disease, received FDA approval in 2006. This marked a turning point in the management of the disease, offering the first effective treatment option.
Impact of ERT on Life Expectancy in Infantile-Onset Pompe Disease
Studies have shown that ERT significantly extends life expectancy in infants with IOPD. Prior to ERT, the median survival for infants with IOPD was less than one year.
With ERT, many infants now survive well beyond their first year, with some living into their teens and adulthood. However, the effectiveness of ERT varies depending on factors such as the age at which treatment is initiated and the severity of the disease.
Early initiation of ERT, ideally before significant muscle damage has occurred, is crucial for maximizing the benefits of treatment. Research indicates that infants who receive ERT within the first few weeks of life have a better chance of survival and improved motor function compared to those who start treatment later.
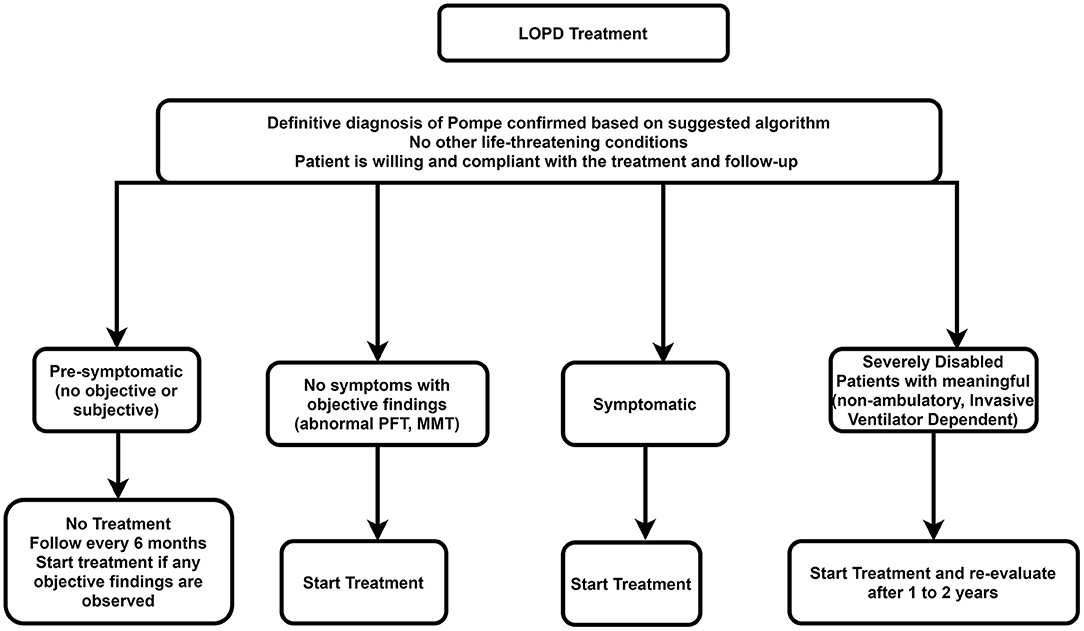
Impact of ERT on Life Expectancy in Late-Onset Pompe Disease
ERT has also demonstrated benefits for individuals with LOPD, although the impact on life expectancy is less dramatic compared to IOPD. In LOPD, ERT can help to slow the progression of muscle weakness, improve respiratory function, and enhance quality of life.
While ERT may not fully reverse the damage caused by glycogen accumulation, it can help to stabilize the disease and prevent further deterioration. Studies have shown that individuals with LOPD who receive ERT have improved survival rates compared to those who do not.
However, the response to ERT in LOPD can vary considerably, and some individuals may experience limited benefits. Factors such as the severity of muscle involvement, the presence of other health conditions, and individual genetic variations can all influence the effectiveness of treatment.
Limitations and Future Directions
Despite the significant advancements in treatment, challenges remain in managing Pompe disease. ERT is not a cure, and individuals with Pompe disease still require ongoing medical care and supportive therapies.
Furthermore, ERT can be expensive, and access to treatment may be limited in some regions. Research is ongoing to develop new and improved therapies for Pompe disease, including gene therapy and other novel approaches.
These emerging therapies hold promise for potentially providing a more effective and long-lasting treatment option. Scientists and pharmaceutical companies are actively working on new drugs to enhance ERT efficacy, exploring alternative enzyme delivery methods, and developing gene therapy approaches that could address the root cause of the disease by correcting the defective GAA gene.
Living with Pompe Disease: A Human Perspective
Beyond the medical statistics, it's important to remember the human aspect of living with Pompe disease. Individuals with Pompe disease and their families face significant challenges, including physical limitations, emotional distress, and financial burdens.
Support groups and advocacy organizations play a vital role in providing information, resources, and emotional support to those affected by Pompe disease. These groups work to raise awareness of the disease, advocate for improved access to treatment, and promote research into new therapies.
The Acid Maltase Deficiency Association (AMDA) and other similar organizations are crucial in connecting patients and families, providing a platform for sharing experiences and offering mutual support.
Conclusion
The introduction of ERT has significantly improved the life expectancy and quality of life for individuals with Pompe disease. While ERT is not a cure, it has transformed the prognosis for both infantile-onset and late-onset forms of the disease.
Early diagnosis and prompt initiation of treatment are essential for maximizing the benefits of ERT. Ongoing research and development efforts are focused on developing new and improved therapies that could further extend life expectancy and improve outcomes for individuals with Pompe disease.
As advancements continue, hope remains high for a brighter future for those living with this challenging condition. Continued collaboration between researchers, clinicians, patients, and advocacy organizations is critical for advancing our understanding of Pompe disease and developing more effective treatments.